
What Is Cystic Fibrosis
Cystic Fibrosis (CF) is the most common, worldwide, inherited disease of the caucasians. It is a genetic disorder, therefore non-communicable, and it’s one of the most common rare diseases. It is caused by a mutation of a gene on the 7th chromosome that encodes the CFTR protein, which regulates the production of body fluids and secretions (sweat, mucus, digestive fluids, etc.).The disease causes thick, sticky and dehydrated mucus in the lungs and other organs, such as the pancreas, liver and intestines. Thick fluids and mucus accumulate in the organs and block the organs in the body. Over time, the organs are damaged, gradually destroyed and lead to failure, creating life-threatening problems such as infections, respiratory failure, malnutrition, etc.

Symptoms
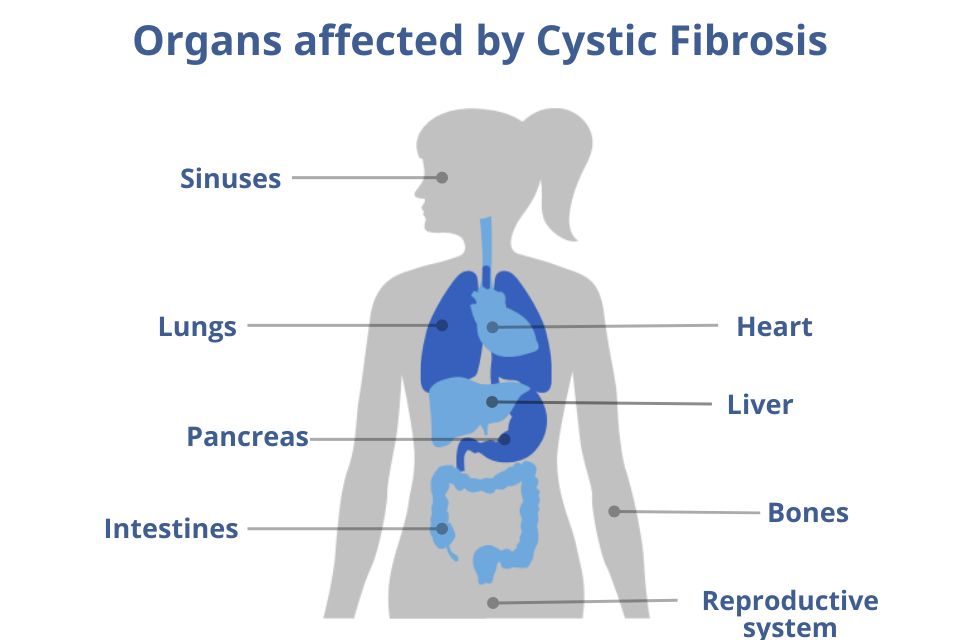
CF is a multisystem, life-threatening disease that affects many organs of the human body, mainly the lungs and pancreas.
The symptoms vary from patient to patient due to the significant heterogeneity of gene mutations, which means that the disease manifests itself differently in each patient, depending on their genotype (genetic profile) and various other factors. The main symptoms concern the respiratory system, the digestive and gastrointestinal system, and the reproductive system.
Salty Sweat
Patients with Cystic Fibrosis, due to the lack or dysfunction of the CFTR protein, have very salty sweat, so in exceptionally high temperatures, especially in the summer months, they may experience episodes of dehydration due to a significant disturbance in electrolyte levels.
Pulmonary Complications
The complications of the respiratory system, which destroy the lungs over time, are considered critical for the survival of the patients, having the overwhelmingly most significant percentage among the causes of mortality of the disease. The symptoms usually presented by patients are continuous persistent cough, thick phlegm, difficulty breathing, wheezing, shortness of breath, bronchospasm, and hemoptysis. Patients also often develop respiratory infections, as thick sticky mucus builds up in the lungs, creating ideal conditions and environment for germs to survive and multiply. Germs and bacteria, such as staphylococcus, pseudomonas aeruginosa, aspergillus, and mycobacteria, usually cause respiratory infections. These microbes typically settle permanently in the lungs of patients and are difficult to eradicate, creating a severe chronic problem. Also, patients often present with nasal polyps and sinusitis.
Gastrointestinal Complications
The accumulation of mucus in the pancreas blocks the passages that carry enzymes produced by the pancreas into the small intestine. Without the specific enzymes, the intestine cannot absorb the necessary nutrients from food, resulting in malabsorption, poor nutrition, and poor physical growth in patients. Patients often experience loss of appetite, abdominal distension, constipation, chronic abdominal pain, chronic diarrhea, greasy and foul-smelling stools, steatorrhea, poor weight gain and growth, and intestinal obstruction, especially in neonates (meconium ileus). Pancreatitis can also occur many times. In the liver, thick mucus may block the bile duct, causing liver disease and severe liver problems such as cirrhosis.
CF Related Diabetes
Due to pancreatic insufficiency, insulin production stops or decreases in many patients. This results in the form of diabetes called Cystic Fibrosis Related Diabetes (CFRD). DMD is a different type of diabetes, which has features of both type 1 diabetes (known as juvenile diabetes) and type 2 diabetes (usually in older and overweight people).
Ιnfertility
The disease affects the patient’s fertility. 97-98% of men with Cystic Fibrosis have obstructive azoospermia and can only father a child through IVF. Women often produce vaginal mucus that is thicker than average, and it usually takes longer to get pregnant naturally.
Other Complications
The disease also affects other body systems, often causing bone diseases such as osteoporosis-osteopenia, joint pain, rheumatoid arthritis, thyroid complications, heart problems such as heart attack or heart failure, and various other complications.
Life expectancy
Cystic fibrosis patients have a low life expectancy.
In the 1960s, the majority of patients died before they were five years old. In the 1980s, only 30% of patients were adults.
Although Cystic Fibrosis is progressive and requires daily care, today many patients are able to attend school, study and work. Their quality of life is significantly improved compared to previous decades.
Thanks to the progress of science, the life expectancy of patients is gradually increasing, and there are patients who have reached 50 years of life.
Based on mathematical models, it is estimated that by 2040 the ratio of children to adult patients will be 25%-75%.
Patients who start receiving early childhood treatment with new innovative therapies are expected to have a life expectancy in the coming decades almost equal to that of healthy people.
How Cystic Fibrosis is Inherited?
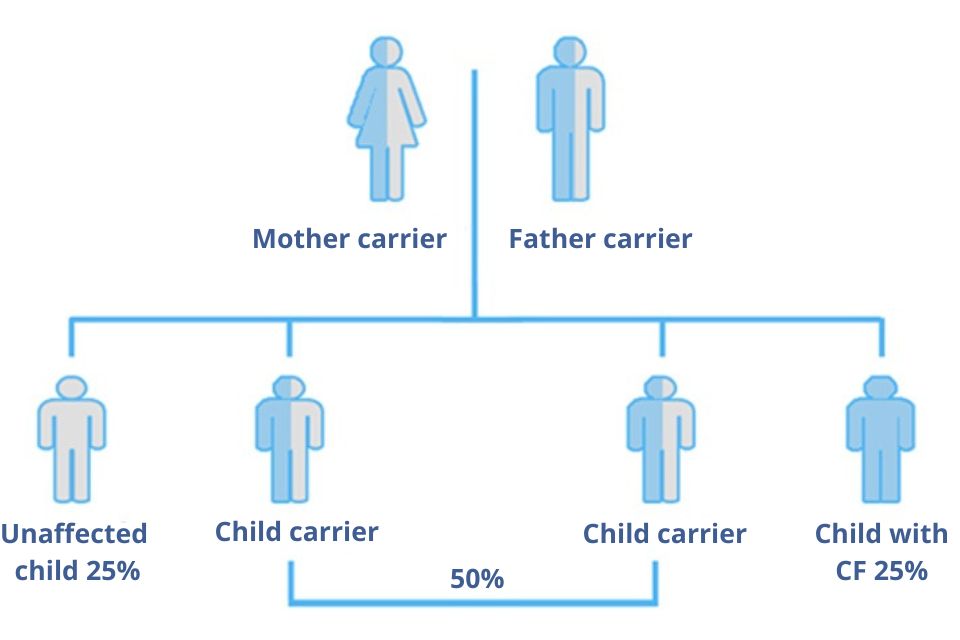
For a child to be born with Cystic Fibrosis both parents must be carriers or patients. Carriers of the disease carry a single abnormal gene and are considered perfectly healthy. In Greece there are more than 500,000 carriers. Cystic fibrosis is inherited like Mediterranean anaemia on the basis of Mendel’s law.
- In the case of two carrier parents, for each child there is a 25% chance of having the disease, 50% of being a carrier and 25% of not even carrying the gene.
- In the case of one parent-patient and one parent-carrier, each child has a 50% chance of having the disease and a 50% chance of being a carrier.
- From a carrier parent and a non-carrier parent, there is no chance of a child being born with the disease, but 50% chance of each child being a carrier.
- From a parent-patient and a non-carrier parent, all children will be carriers.
- Two parents who are both patients can obviously only have children who will have the disease.
Frequency
It was not until 1989 that the gene associated with Cystic Fibrosis was identified, and to date, more than 2,100 mutations of the CFTR gene have been identified.
The most frequent gene mutation observed in Greece is the one characterized as F508del or ΔF508, which is considered one of the most severe mutations in symptoms. It has an incidence of approximately 70% in North America, 70-80% in northern European countries, and 30-54% in southern European countries, while in our country, its frequency is 53% of patients. In Greece, the number of patients exceeds 800.
It is estimated that every year 1/2,000-2,500 children are born with Cystic Fibrosis and a total of 40 children per year in Greece. In Greece the 5% of the general population, about 500,000 people, is considered to be carriers of Cystic Fibrosis.
In Greece the 5% of the general population, about 500,000 people, is considered to be carriers of Cystic Fibrosis.
Although in Greece and other Mediterranean countries, Mediterranean Anemia shows twice the percentage of carriers (8-10%), due to the information and the extensive program of prenatal control that has been implemented in the last 20 years, the births of sick children do not exceed five (5) every year. Therefore, from the point of view of the birth of new sufferers, Cystic Fibrosis is by far the most widespread hereditary disease in Greece.
The timeline
of Cystic Fibrosis
The first mention of the disease appears in 1705 in a folklore book, stating that "a child with a salty taste is bewitched."
In a book of Children's Songs and Games published in Switzerland, the medieval saying is included: "Woe to the child whose forehead is kissed with a salty taste because he is bewitched and will soon die."
Dr. Dorothy Andersen first describes the mysterious disease that kills children as "Cystic Fibrosis of the Pancreas."
A modern comprehensive treatment plan for patients is proposed for the first time
The first double lung transplant in patients with Cystic Fibrosis is performed.
Cystic Fibrosis gene discovered in Canada by Lap-Chee Tsui, Francis Collins, and Jack Riordan.
First inhaled mucolytic drug designed explicitly for Cystic Fibrosis approved.
The first inhaled antibiotic for Cystic Fibrosis is approved.
The first meeting of the European Cystic Fibrosis Community (ECFS) physician group with the EurocareCF research group to create the European CF Patient Registry takes place.
The first innovative CFTR Modulator treatment that targets the causes of the disease is approved.
The 4th CFTR Modulator treatment is approved by FDA for 90% of CF patients worldwide with extraordinary results.
Evidence on the impact of covid-19 on CF patients is being studied and published.
Cystic Fibrosis Gene Therapy and Bacteriophage Therapy Clinical Trials Underway.