From the first moment of diagnosis, patients with CF are followed for life in a specialised Cystic Fibrosis centre and undergo a combination of time-consuming treatments on a daily basis. Patients’ daily struggle requires dedication and discipline. For patients in the final stages of respiratory failure, the only chance of survival is a lung transplant.
Symptomatic treatments
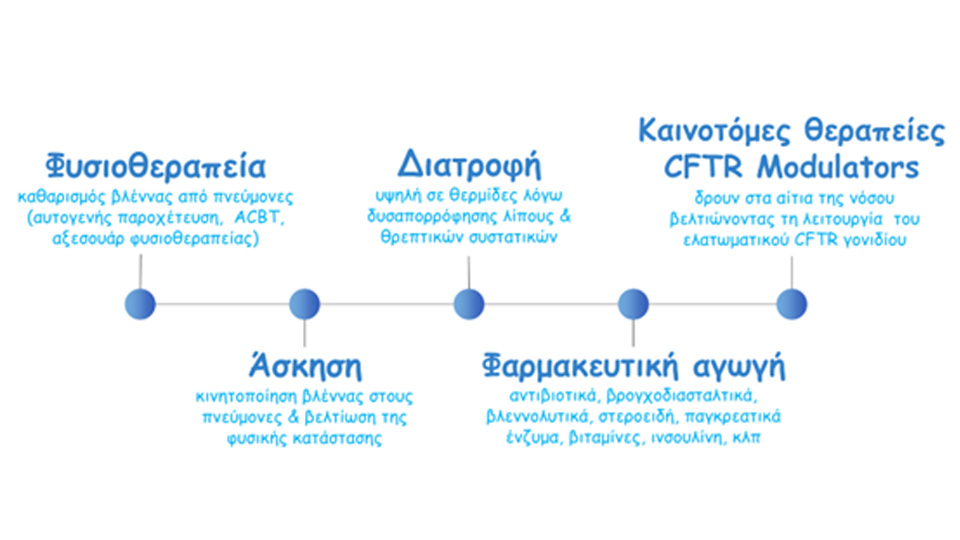
Cystic fibrosis patients follow a combination of time-consuming daily treatments that last 3-4 hours each day and follow a strict medical care protocol. They also require regular hospitalisation at a specialist CF centre.
The main aim of the treatments is to prevent and treat respiratory infections that lead to respiratory failure, but also to relieve the symptoms of the disease. They also aim to keep the lungs in particular, but also the patient’s body in general, in the best possible condition.
Thoracic respiratory physiotherapy
Physiotherapy helps to mobilise and remove the thick secretions and sputum that accumulate in the airways of patients’ lungs. It is done on a daily basis and its frequency and duration vary with each patient. There are various techniques that are used, such as autogenous drainage, active cycle breathing techniques, positive expiratory pressure with devices such as PEP mask, flutter, aerobics, acapella, etc.
Exercise
In combination with physiotherapy, frequent exercise helps to mobilise the mucus in patients’ lungs, strengthen their musculoskeletal system and improve their physical condition.
Inhaled drugs in a nebuliser
Patients receive daily medication through inhalation into a nebuliser, which is a device that turns drugs into a cloud. These drugs can be either bronchodilators, mucolytics to break down mucus (Dornase alpha), or antibiotics (tobramycin, azreonam, colistin, levofloxacin, etc.) to keep the microbial load in the patients’ lungs low.
Nutrition & pancreatic enzymes
Patients need to follow a diet rich in calories, fat and protein in order to maintain a good weight and good nutrition. To improve the absorption of nutrients in food, they take daily pancreatic enzymes with every meal or snack. Enzymes are essential for breaking down food into components that can be absorbed by the body for normal digestion.
Intravenous treatments
Due to exacerbations of the disease, patients are often hospitalized for about 2-3 weeks in specialized Cystic Fibrosis Centers, where they receive intravenous treatments to treat respiratory infections or other complications of the disease.
Other treatments
Due to malabsorption many patients take ABDEK vitamin preparations, but also dietary supplements. Also some patients are given insulin to treat Cystic Fibrosis Diabetes Mellitus. Various treatments are used to treat chronic sinusitis, such as nasal washes with sea water, antihistamines, corticosteroids and antibiotics. Nasal polyps can be treated surgically, but recurrences are very common. Patients are also recommended to hydrate frequently and eat salty foods, especially during the summer months, to avoid dehydration.
Innovative CFTR Modulators treatments
Innovative CFTR modulator therapies (CFTR Modulators) act on the causes of the disease. They improve the function of the defective CFTR gene, which causes Cystic Fibrosis, and “freeze” the disease, significantly slowing its progression.
Thanks to innovative CFTR modifier therapies, the health and quality of life of patients with Cystic Fibrosis has improved significantly in recent years, and their survival rate is gradually increasing.
The first innovative treatments
In 2012, the first innovative CFTR Modulator therapy Ivacaftor was launched under the trade name Kalydeco, which targets the causes of the disease and is administered to a small number of patients, as it targets a limited number of gene mutations. A few years later, the next two CFTR treatments lumacaftor/ivacaftor (Orkambi) and tezacaftor/ivacaftor (Symkevi) were released, targeting a larger number of gene mutations. These treatments can be given to patients who are homozygous for the most common gene mutation F508del, but they do not have the same efficacy as the first treatment.
The revolutionary triple combination therapy
Gene therapy
Gene therapy aims to replace the defective CFTR gene in the airway cells of patients with Cystic Fibrosis and is aimed at all patients, regardless of their mutations. At present, gene therapy is in the clinical trials stage, with further developments expected in the coming years.
Lung transplantation
For patients with Cystic Fibrosis who are not eligible for an innovative CFTR modifier therapy or do not have the expected results from innovative therapies and are in the final stages of respiratory failure, the only survival option is lung transplantation.
The transplant is carried out if they are approved for inclusion on the transplant list. To be included on the list, a patient must meet certain criteria, which are established through a series of tests during pre-transplant screening. When a suitable graft is found in time, the transplantation procedure takes place. Before and after the transplantation, the patient is given medication to prevent the rejection of the transplant.
The monitoring of the transplant patient never stops and is done every three or six months, depending on the patient’s condition. Several times hospitalisation is needed in case of any deterioration of respiratory function or if any symptoms from the respiratory system occur. A major issue facing a transplant patient is graft rejection, as well as frequent and serious infections. Based on the statistics, the median survival for lung transplantation in patients with Cystic Fibrosis is longer compared to other respiratory diseases, due to the young age of Cystic Fibrosis patients transplanted.
More about lung transplants on the website of the National Transplantation Organisation (NTO)
here.
Transnational transplant agreement with Austria
Until 2014, there was no stable long-term programme for lung transplantation in Greece. With pressure from the Panhellenic Cystic Fibrosis Association in cooperation with the State and the competent bodies, a transnational cooperation agreement between the National Transplantation Organisation (EOM) and the Austrian Transplant Centre (AKH) was signed in 2014. Thanks to this agreement, more than 25 patients with Cystic Fibrosis, as well as patients with other respiratory diseases, were able to undergo successful lung transplantation in Austria from 2015 to 2019.
National Lung Transplant Programme
In June 2019, the transnational cooperation agreement between Greece and Vienna, under which Greek patients were referred to the AKH hospital for lung transplantation, expired. Since 2019, the National Lung Transplant Program has been launched in Greece at the Onassis Cardiosurgery Center with the support of the Onassis Foundation. This is where all Greek patients who need a lung transplant are referred to. Between 2020 and November 2022, 18 lung transplants were performed in the Greek programme. The first two successful lung transplants in Cystic Fibrosis patients were performed in 2022.
Organ donation
In Greece, organ donation rates are low. Lungs in particular require even more effort than other organs, as they are a fragile and sensitive graft. The timely identification of potential donors in hospital ICUs and the timely procurement of organs is a difficult process, which requires a well-organised health system with a sufficient number of qualified scientists. To become an organ donor, a person must be certified in the ICU as having brain death, which is an irreversible condition. Organ donation is the ultimate act of love and altruism, as one donor can save eight of our fellow human beings.
Campaign "Unlimited Breath - Be a Life Donor"
The Panhellenic Cystic Fibrosis Association participates in the national effort to increase the number of organ donors in Greece with the successful campaign “Breath unlimited – Be a life donor”. The Association has created the campaign since 2021, under the auspices of the National Transplantation Organization and the Onassis Cardiac Surgery Center, with the aim of raising public awareness about organ donation and transplantation.
Campaign communication actions: murals in major hospitals across the country with stories of real Greek transplanted patients with CVI who won a second chance at life, TV video spots, awareness concert.
Honorary Supporter of the Campaign is the President of the Republic.
The campaign has received the Gold Healthcare Award 2024 and Silver Healthcare Award 2021, as it has helped raise awareness of organ donation and transplantation. Indicatively in the midst of covid-19 in 2020-2021 it helped to triple organ donor registrations, with 631 new registrations on the National Transplantation Agency’s Organ Donor Register.
Become an organ donor and see the campaign’s actions here.
